Por extraño que parezca, las enfermedades raras tienen algunos rasgos en común; uno puede ser el origen genético; otro, la afectación enzimática. Dieciséis años después de que Gaucher identificara un déficit enzimático con la enfermedad a que dio nombre, el alemán Johannes Fabry descubrió una nueva rara enfermedad caracterizada por la perentoriedad de la alfa-galactosidasa A.
En clínica se ha bautizado también esta enfermedad como angioqueratoma difuso, déficit de ceramida trihexosidasa, déficit de alfa-galactosidasa A, lipidosis glucolipídica, angioqueratoma corporal difuso, enfermedad de Anderson (un médico inglés que disputó a Fabry la identificación del déficit enzimático) o lipidosis hereditaria distópica. La enfermedad de Fabry es un trastorno hereditario que afecta más a los hombres que a las mujeres. Se calcula que uno de cada 40.000 varones padece esta enfermedad, mientras que la prevalencia en la población general es de un solo caso por cada 117.000 personas. Clínicamente el primer síntoma suele ser el dolor, en forma de crisis agudas y lacerantes de dolor abdominal difuso, refractario al tratamiento, que pueden duran dos ó tres semanas o incluso más y desaparecer bruscamente. Las crisis dolorosas pueden desencadenarse por fiebre, fatiga, ejercicio, estrés, o cambios de temperatura. Estas crisis son más comunes en la infancia y una vez que el paciente alcanza la edad adulta, dichas crisis pueden llegar a desaparecer, o por el contrario, el dolor puede agravarse a medida que pasan los años.
Manifestaciones clínicas
Se han descrito dos formas clínicas de la enfermedad. La más clásica, que aparece en el 90-95% de los casos, con total ausencia de actividad enzimática y evolución progresiva con acroparestesias, angioqueratoma y afectación de los órganos diana por acúmulo de lisosomas. Y otra poco frecuente que aparece a partir de los 45 años, con una actividad enzimática residual, sin acúmulo de lisosomas y en la que el órgano principalmente afectado es el corazón.
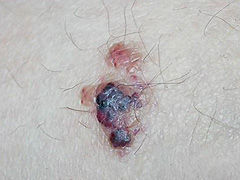
Las manifestaciones clínicas más frecuentes son alteraciones de la piel, angioqueratomas difusos que aparecen en pacientes jóvenes (10-20 años). Las lesiones aparecen con preferencia en los flancos, región infraumbilical y genitales. Este cuadro puede acompañarse de un aspecto facial de ‘cara basta’, con cejas muy prominentes. También pueden presentarse alteraciones oculares del tipo de una córnea vertiginata (nebulosa corneal excéntrica) y disfunciones en los vasos de la retina. Las alteraciones viscerales aparecen alrededor de los 20 años de edad y son debidos a la acumulación de lisosomas. En el riñón, los lisosomas son responsables de un fracaso renal que requiere diálisis o trasplante. En el corazón, aparecen arritmias, cardiomegalia (aumento del tamaño del corazón), insuficiencia mitral, infarto agudo de miocardio o cardiomiopatía hipertrófica (engrosamiento de las paredes del corazón).
Las manifestaciones más frecuentes en pacientes jóvenes son alteraciones de la piel, con preferencia en los flancos, región infraumbilical y genitales
Las alteraciones del sistema nervioso central (sistema formado por el encéfalo y la médula espinal) abarcan signos de vértigo, tinnitus (repiqueteo percibido en uno o ambos oídos), cefalea, un cierto grado de sordera, ictus y deterioro cognitivo. Otros síntomas asociados con la enfermedad de Fabry son bronquitis crónica, disnea, calambres en las piernas, diarrea insidiosa, osteoporosis (desmineralización esquelética generalizada) y retrasos en crecimiento y pubertad.
La genética manda
La enfermedad de Fabry se ha definido como un error congénito del catabolismo de los glucoesfingolipidos. Los genes que se transmiten de padres a hijos no sólo se refieren a la estatura, el color de la piel, del cabello o los ojos. El padre, la madre o ambos progenitores pueden ser portadores de un gen defectuoso (mutante) que puede causar una enfermedad. El gen que causa la enfermedad de Fabry se encuentra localizado en el cromosoma X, por lo que, aunque la enfermedad se manifieste primordialmente en varones, también las mujeres pueden experimentar sus síntomas.
Cuando una persona hereda el gen mutante de la enfermedad de Fabry, su cuerpo no puede producir suficientes cantidades de alfa-galactosidasa A, requerida para eliminar un lisosoma (estructura celular que funciona como unidades digestivas elementales de las sustancias grasas) denominado globotriaosilceramida. Sin la enzima pertinente, este lisosoma se acumula de forma perniciosa en las células de los vasos sanguíneos, hígado, corazón, piel y cerebro. La acumulación persistente de globotriaosilceramida en los tejidos celulares puede conducir eventualmente a problemas graves y muerte.
Lo que caracteriza a la enfermedad de Fabry, con respecto a otras enfermedades graves de origen genético es su dificultad diagnóstica. Por lo tanto, las personas enfermas pueden pasar largos periodos sin saber realmente qué padecen. Los médicos se apresuran a dar con claves eficaces de identificación, puesto que mientras más largo sea el periodo transcurrido sin tratamiento, más probable es la aparición de lesiones irreversibles en los órganos y tejidos del cuerpo.
La enfermedad de Fabry se hereda como un rasgo dominante ligado al cromosoma X. El gen que controla la alfa-galactosidasa A se identificó la pasada década y se localiza en el brazo largo del cromosoma X (Xq22.1). Existen más de 160 mutaciones descritas, que en el 76% de los casos constan de una simple sustitución de nucleótidos. No se aprecia correlación entre la expresión genotípica y la expresión fenotípica de la enfermedad, siendo tan elevada la variabilidad genética que se podría decir que cada familia tiene su propia mutación. Si la madre es la portadora y el padre no tiene la enfermedad, hay un 50% de probabilidad que una hija sea portadora de la enfermedad.
Pacientes y familiares de la enfermedad de Fabry disponen en España de una Asociación para las Deficiencias que afectan al Crecimiento y al Desarrollo (ADAC), además de la Federación Española de Asociaciones de Enfermedades Raras (FEDER) y la European Organization for Rare Disorders (EURORDIS).
Tratamiento
En los últimos años se ha empezado a utilizar alfa-galactosidasa A en terapia génica, aplicando la enzima de forma intravenosa. Una vez dentro del organismo, la alfa-galactosidasa A disuelve los cúmulos de lisosomas y mejora la función celular. La infusión de alfa-galactosidasa A intravenosa se aplica ya cada dos semanas en los protocolos de algunos centros estadounidenses, pese a que el tratamiento como tal no ha sido aún aprobado por la Food and Drug Administration (FDA).
Actualmente se experimenta con ratas de laboratorio en las que no se implanta ya la enzima, sino el gen responsable de su producción, por lo que se espera que en un futuro bien cercano esta modalidad terapéutica permita mejorar sensiblemente la calidad de vida de los enfermos.
El diagnóstico de la enfermedad de Fabry es muy difícil, ya que la mayoría de sus síntomas son inespecíficos y puede no confundirse con muchas enfermedades: fiebre reumática, colitis ulcerosa, síndrome de Raynaud, gota, encefalitis diseminada y enfermedad de Churg Strauss. El diagnóstico de sospecha basado en la clínica y se confirma con la determinación de la actividad de la enzima alfa-galactosidasa A en sangre o piel.
La primera clave de sospecha clínica es un dolor que afecta principalmente a manos y pies, la aparición de angioqueratomas, pérdida de sensación al frío o al calor en las partes distales de las extremidades y presencia de opacidades en la córnea. Estos signos bastan para detectar anormalidades en plasma, leucocitos, lágrimas o tejidos obtenidos por medio de una biopsia que permitan determinar la deficiencia de alfa-galactosidasa A.
El diagnóstico prenatal se puede hacer también por medio de amniocentesis. El examen de orina mide los niveles de actividad enzimática y puede resultar igualmente orientativo. Un diagnóstico precoz de la enfermedad de Fabry permitirá al médico iniciar con prontitud el tratamiento y controlar mejor sus síntomas, previniendo problemas de salud adicionales.
Algunas personas padecen la enfermedad de Fabry, pero únicamente con síntomas cardiacos; estas personas tienen una cantidad más elevada de enzima, por lo que pueden escapar al diagnóstico precoz, y presentan cuadros de insuficiencia cardiaca, y/o latidos irregulares. Sólo un médico avezado dará con la oportunidad del diagnóstico a partir de una biopsia de corazón. La tríada clínica característica que define la enfermedad de Fabry es neuropatía distal dolorosa, angioqueratomas (verrugas decoloradas en la piel, que se agrupan en forma de racimos), e hipohidrosis (disminución o ausencia de sudoración).